Prediction of native binding modes (NBM)
| Assay |
NBM ranked first |
NBM within the top 5 |
| Native docking |
55 % |
64 % |
| Cross docking |
26 % |
44 % |
Using predicted binding modes
Once your job is terminated, you will receive an e-mail with a link to a reference complex and predicted binding modes. They can be converted to your favorite format, or used directly. The ViewDock plugin of UCSF Chimera is very convenient to explore the predicted binding modes.
For experimented users, CHARMM PSF/CRD/RTF/PAR files are also provided for subsequent calculations.
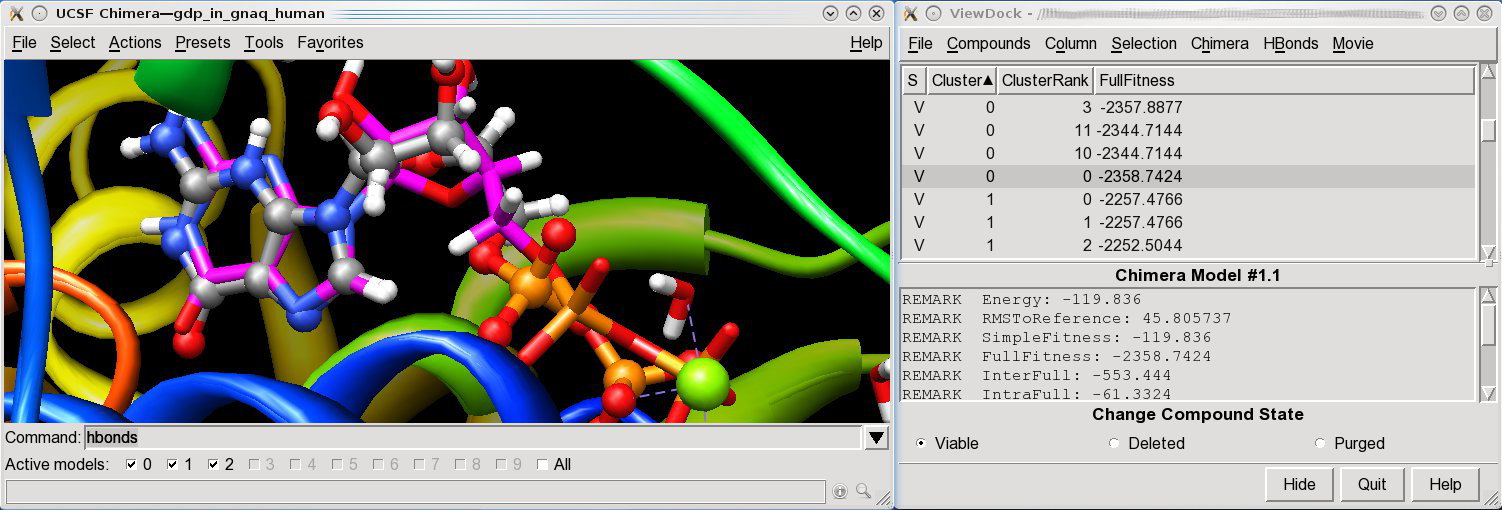
Docking of
GDP in
GNAQ_HUMAN,
a therapeutic target in oncology.
The predicted binding mode (magenta/sticks) is superimposed to the
X-ray binding mode (ball and sticks)
(download the chimera session ).
Upload a PDB or a ZIP file
The structure of the target protein can be uploaded either:
as a ZIP file containing your target protein in the CHARMM format (PSF, CRD and, if needed, extra RTF/PAR).
as a PDB file
Search for a target protein
You can search for targets using a PDB code, a name, an aminoacid sequence, or even load it from a URL.
PDB codes, names and aminoacid sequence will be looked for in the PDB database.
Names will also be searched for in our database of clean targets.
Load a target protein from a URL
You can also load a file from a URL, provided that it is either:
- a PDB file
- a ZIP file containing your target protein in the CHARMM format (PSF, CRD and, if needed, extra RTF/PAR).
Upload a file
A success rate >80% can be achieved with drug-like ligand with less than 15 free dihedral angles.
The structure of the ligand can be uploaded either:
as a MOL2 file with all hydrogens and 3D coordinates. Check atom chirality,
and adjust protonation states according to your needs (e.g. carboxylate groups are usually deprotonated at physiological pH), and make sure that it has a correct topology (we recommand UCSF Chimera, OpenBabel, MarvinSketch, XDrawChem, ChemDraw).
as a ZIP file containing files in the CHARMM format (PDB/RTF/PAR).
If you upload a Mol2 file, the Merck Molecular Force Field (MMFF) will be used to derive CHARMM parameters for your ligand, as follows:
dihedral angle terms will be taken as is
only the harmonic part of the bond, angle and improper terms are retained
charges will be taken from MMFF
van der Waals parameters will be taken from the closest atom type in CHARMM22.
For consistency with CHARMM, atoms with identical names will be renamed. For more, please check the SwissParam website.
The Click2Drug resource portal contains a list of Mol2 related-tools.
Search for a ligand
A success rate >80% can be achieved with drug-like ligand with less than 15 free dihedral angles.
You can search for ligands using a ZINC accession number (AC), its name, or its category.
ZINC AC and names will be looked for in the ZINC database.
Names and categories (scaffolds or sidechains) will be searched for in our database of 58 compounds consisting of 27 scaffolds and 31 sidechains. See here and here for further details.
Load a ligand from a URL
You can also load a file from a URL, provided that it is either:
a MOL2 file with all hydrogens and 3D coordinates. Check atom chirality,
and adjust protonation states according to your needs (e.g. carboxylate groups are usually deprotonated at physiological pH), and make sure that it has a correct topology (we recommand UCSF Chimera, OpenBabel, MarvinSketch, XDrawChem, ChemDraw).
a ZIP file containing files in the CHARMM format (PDB/RTF/PAR).
Before moving on, make sure that the protonation states are reasonable, since they have a big impact on the docking outcome.
Docking type
Here are the success rates observed on the 260 complexes of S3DB, for native blind docking:
| Docking type |
SR=0 |
SR<5 |
CPU time |
| Very fast |
49 % |
62 % |
13 min |
| Fast |
54 % |
63 % |
19 min |
| Accurate |
55 % |
64 % |
24 min |
| SR=0 |
: Success rate considering the top pose |
| SR<5 |
: Success rate considering the top five poses |
The selected docking time determines the number of sampled binding modes, the number of minimization steps that are performed to relax the ligand, and the number of putative binding modes that are sent back to you.
If your ligand has less than 15 rotatable bonds and/or is likely to fit exactly into a binding pocket of your target, the very fast and fast modes are likely to be sufficient.
Definition of the region of interest (ROI)
These fields allow you to define a region in space to which the docking will be restricted (local docking). The box should encompass the putative binding site as well as alternative sites, so that binding modes can have a chance to be predicted outsite of the former, otherwise the docking has little sense.
The region in space will be centered on the point (X center, Y center, Z center), and its maximum dimensions will be (X size, Y size and Z size).
If you want to perform a blind docking, just leave all these fields blank.
Flexibility of the protein
This field allow you to choose the amount of flexbility of the protein that should be considered during the docking.
If the distance is set to 0Å, the protein will be considered rigid.
If the distance is set to 3Å, the side chains that are in close contact with the ligand in its reference binding mode will be co-optimized during the docking.
If the distance is set to 5Å, the side chains that are a bit further from the ligand in its reference binding mode will be co-optimized during the docking.
Although this feature already led to successful stories (e.g. PMID:17200111), it has not been extensively tested yet.
Prediction of native binding modes (NBM)
| Assay |
NBM ranked first |
NBM within the top 5 |
| Native docking |
55 % |
64 % |
| Cross docking |
26 % |
44 % |
Using predicted binding modes
Once your job is terminated, you will receive an e-mail with a link to a reference complex and predicted binding modes. They can be converted to your favorite format, or used directly. The ViewDock plugin of UCSF Chimera is very convenient to explore the predicted binding modes.
For experimented users, CHARMM PSF/CRD/RTF/PAR files are also provided for subsequent calculations.
Docking of
GDP in
GNAQ_HUMAN,
a therapeutic target in oncology.
The predicted binding mode (magenta/sticks) is superimposed to the
X-ray binding mode (ball and sticks)
(download the chimera session ).